Кажется я созрел до реальной работы над этой задачей, готов скоро приступить.
Сейчас я сделал программу, которая запускает программу xtb, та делает single point расчёт энергии и её градиентов, далее моя программа читает файл xtb и итерация повторяется. Что-то близкое сделал Артём Оганов в своём Успехе, как я понимаю.
Текущий алгоритм оптимизации крайне примитивный: к координатам атомов добавляются градиенты, умноженные на какой-то коэффициент. Если на следующем шаге энергия получилась ниже – система возвращается к предыдущим координатам, и этот коэффициент уменьшается.
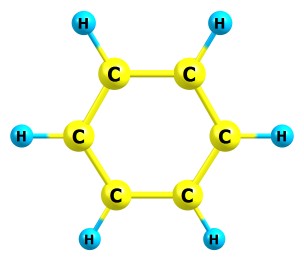
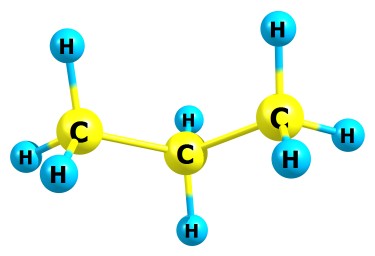
Интересно сейчас получилось - такая молекула, как бензол, оптимизируется нормально, в то время как например пропан очень долго (если у пропана повернуть метильные группы вокруг связи C-C, они никак не соптимизизируются в правильное положение). Предлагаю догадаться, почему так получается. Вот структуры этих молекул:
Итак, каким у меня будет алгоритм: я сделаю кастомный ММ функционал, и его параметры будут динамически подгоняться под квантовохимический расчёт. Я не хочу изучать литературу по молекулярной механике, поскольку, как мне кажется, у меня лучше получится сразу придумать что-то своё, учитывая что я уже 20 лет занимаюсь структурной и квантовой химией. В моём алгоритме будут следующие параметры: три коэффициента для потенциала Морзе для каждой связи, и по два параметра для каждого атома, определяющих значения коэффициентов для потенциала Леннарда-Джонса с этим атомом, точнее определяющих как бы способность атома притягивать другие атомы с энергией, обратно пропонциональной шестой степени от расстояния, и отталкивать их с энергией, обратно пропорциональной двенадцатой степени. Понятно ли кому-то, о чём я говорю?
Возьмём для примера молекулу толуола:
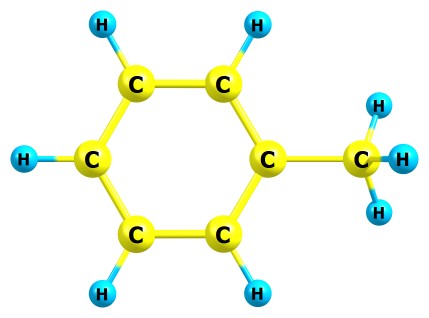
Здесь 15 атомов и 15 связей. Значит суммарное число параметров будет

. Эти параметры будут фиттиться на каждом шаге оптимизации.
Когда будет посчитана первая точка, расчёт даст только градиенты, т.е. этот расчёт покроет

степеней свободы (из них ещё 6 избыточных). Это значит, что фиттить ММ параметры неосторожно будет нельзя, иначе система улетит. Если же расчёт дойдёт до точки минимума и посчитаются частоты (вторые производные), он даст

чисел, этого разумеется хватит для фита, и надо будет добавить ещё какие-то параметры, чтобы не недофитилось (недоиспользование данных расчёта частоты). Продолжение следует.